
Liver disease from microbial infection is a major public health concern. Hepatitis C virus (HCV) infects the liver to cause liver disease, liver cancer and death. Our studies aim to define the virus and host components that control HCV infection outcome and liver disease. These studies in particular, will complement projects in the Gale laboratory that are focused on developing improved HCV culture systems and understanding the role of the viral proteins and host interferon stimulating genes in controlling HCV.
The RIG-I pathway and HCV infection
Steps 1-6 denote HCV entry, PAMP recognition, translocon assembly, RIG-I/MAVS binding, IRF-3/NF-κB activation, and induction of innate immune gene expression.
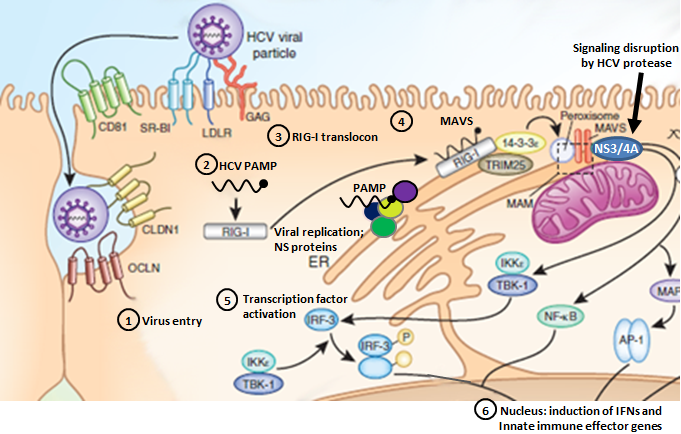
- HCV Entry:
HCV enters the host cell through a coordinated pathway of sequential co-receptor interactions that has not been entirely elucidated. The complex orchestration of HCV co-receptors during HCV binding and entry suggests that dysregulation of the co-receptor interaction process offers a prime therapeutic strategy to suppress HCV infection. Our results define IFITm1 as an innate immune effector gene that imparts antiviral actions of IFN against HCV through the nvoel action of disrupting viral co-receptor interactions. Modified from Wilkins et al., 2013
-
PAMP Recognition:Pathogens are sensed by host pattern recognition receptors (PRRs) that recognize molecular motifs (Pathogen-Associated Molecular Pattern). Two major receptor systems sense the presence of viral infection to mount an immune response: toll-like receptors (TLRs) 3, 7, 8, and 9 are major PRRs that respond to different types of viral nucleic acids, and more recently, retinoic acid inducible gene-I (RIG-I)-like receptors (RLRs), helicases including RIG-I and MDA-5 (melanoma differentiation-associated gene 5), have been identified as cytosolic receptors for intracellular dsRNA sensing. Modified from Stone et al., 2013
- Translocon Recognition:
RIG-I is a cytosolic pathogen recognition receptor that initiates immune responses against RNA viruses. Upon viral RNA recognition, anti-viral signalling requires RIG-I redistribution from the cytosol to membranes where it binds the adaptor protein, MAVS (mitochondrial antiviral-signaling protein). We have identified the mitochondrial targeting chaperone protein, 14-3-3ε, as a RIG-I binding partner and essential component of a translocation complex or "translocon" containing RIG-I, 14-3-3ε, and the TRIM25 ubiquitin ligase. The RIG-I translocon directs RIG-I redistribution from the cytosol to membranes where it mediates MAVS-dependent innate immune signalling during acute RNA virus infection. Modified from Liu et al., 2012
- RIG-I/MAVS Binding:
MAVS resides on mitochondria and peroxisomes [in the host cell], but how its signaling is coordinated among these organelles has not been defined. We have shown that a major site of MAVS signaling is the mitochondrial-associated membrane (MAM), a distinct membrane compartment that links the endoplasmic reticulum to mitochondria. During RNA virus infection, RIG-I is recruited to the MAM to bind MAVS. Dynamic MAM tethering to mitochondria and peroxisomes then coordinates MAVS localization to form a signaling synapse between membranes. Importantly, the hepatitis C virus NS3/4A protease, which cleaves MAVS to support persistent infection, targets this synapse for MAVS proteolysis from the MAM, but not from the mitochondria, to ablate RIG-I signaling of immune defenses. Thus, the MAM mediates an intracellular immune synapse that directs antiviral innate immunity. Modified from Horner et al., 2011
- IRF-3/NF-κB Activation:
NF-κB is considered a central positive regulator of the inflammatory response, and its role in the induction of genes such as those for tumor necrosis factor alpha (TNF-α), interleukin-8 (IL-8), and IL-1β has been well characterized. Virus-induced PRR signaling leads to phosphorylation and ubiquitin-mediated proteasomal degradation of these repressor proteins, allowing activated NF-κB to translocate into the nucleus and bind to the promoters of proinflammatory genes. Activation of the downstream effector molecule IRF3 can also lead to the induction of a variety of proinflammatory cytokines, including IRF family members, other signal transudction factors and specific Interferon Stimulated Genes (ISGs) with potent viral regulatory activity. Modified from Horner & Gale, 2013 and Brownell et al., 2014
- Induction of Innate Immune Gene Expression:
Antiviral defenses are triggered by pathogen recognition and signaling that induces IFN and drives the expression of hundreds of ISGs encoding innate immune effectors that impart control of virus replication and spread. Although direct antiviral ISG action is important to control HCV replication, full restriction of HCV infection is mediated by additional actions of innate immune signaling amplification through IRFs and other virus-response pathways. Modified from Horner & Gale, 2013